Main Article by Beatriz Alvarez from Laboratorio de Enzimología, Facultad de Ciencias, Universidad de la República, Montevideo, Uruguay
Correspondig author e-mail: beatri@qualquerz.alvarez@fcien.edu.uy
Highlights. Hydrogen sulfide (H2S/HS–) can be formed in mammals and trigger physiological effects. It can react with metal centers and oxidized thiol products such as disulfides (RSSR) and sulfenic acids (RSOH). Reactions with oxidized thiol products form persulfides (RSSH/RSS–). Persulfides are more acidic and nucleophilic than thiols and, unlike thiols, they can also be electrophilic. They are also good reductants. The modification of critical cysteines to persulfides has been proposed to transduce the effects of H2S.
Hydrogen sulfide, a molecular relic with novel roles
Hydrogen sulfide (H2S) is a gas with the characteristic smell of rotten eggs. Its structure is comparable to water, but due to the lower electronegativity of sulfur with respect to oxygen, H2S is less polar than water and has low intermolecular forces, so it is a gas at room temperature. It is a weak diprotic acid with pKas of 7 and 17. Thus, at physiological pH, 70 % is available as HS– and 30 % as H2S.
H2S was present in the environment where life arose, and it was likely part of the first energy-yielding metabolic process. Today, microorganisms can use or form H2S in various ways. For example, lithotrophs use H2S as electron donor to respiratory chains. For several species including mammals, H2S is toxic because it inhibits cellular respiration. This toxicity has been linked to massive extinction events in the history of life on Earth, such as the Permian–Triassic extinction when more than 96 % of marine species were killed. Nevertheless, in the nineties it was recognized that H2S can be synthesized in mammals and that it can exert several physiological effects with potential health benefits. Numerous effects have been reported, including neuromodulation and attenuation of ischemia-reperfusion injury. Yet, the molecular bases of the observed effects are typically unclear.
The formation of H2S in mammals relies on enzymes of sulfur amino acid pathways: cystathionine beta-synthase, cystathionine gamma-lyase and mercaptopyruvate sulfur transferase. Intestinal bacteria can also synthesize it. H2S can be measured in tissues; the reported values have decreased with time, as analytical methods have improved, and are in the tens of nanomolar range. H2S can rapidly traverse membranes. Mitochondria are the main site for H2S catabolism. By the action of several enzymes, H2S fuels electrons to ubiquinone and is oxidized to thiosulfate (S2O32–), sulfite (SO32–) and sulfate (SO42–). Besides, H2S is an inhibitor of cytochrome c oxidase.
H2S (rather, HS–) is nucleophilic, which means that it can donate a pair of electrons to form a bond. In this regard, it is analogous to thiols (RSH), although H2S is a slightly weaker nucleophile and the products formed are different. One of the possible reactions of H2S in biological contexts is with metal centers. For example, H2S can bind to ferric heme. The binding can be reversible, as in the case of hemeproteins that transport H2S in invertebrates, or the iron can be reduced by H2S. In some cases, sulfheme is formed, a green variant in which the heme ring is covalently modified. Reactions of H2S with oxidants derived from the partial reduction of oxygen (e.g. H2O2) can occur, but direct scavenging is unlikely to explain antioxidant effects. H2S is also able to react with oxidized derivatives of thiols such as disulfides (RSSR) or sulfenic acids (RSOH). In these reactions, persulfides (RSSH/RSS–) are formed (Eqs. 1 and 2).
Importantly, H2S cannot react with reduced thiols. In fact, another reaction that forms persulfides is that of thiols with products of H2S oxidation, such as polysulfides (HSnSH).
Persulfides, possible transducers of the biological effects of H2S
Protein and non-protein persulfides are detected in biological samples and can be formed in thiols through several pathways, some of which are dependent on H2S. The observation that H2S can trigger physiological effects and that it can yield persulfides has led to the proposal that the signaling effects of H2S may in part involve persulfides. Thus, the posttranslational modification of critical protein cysteines to persulfides could unleash downstream effects of H2S. Besides this role in signaling, persulfides participate in several biosynthetic pathways, such as in the assembly of iron-sulfur clusters. In addition, there exist enzymes able to produce (e.g. sulfide quinone oxidoreductase), transfer (e.g. rhodanese) or react (e.g. persulfide dioxygenase) with persulfides.
Persulfides have a very rich biological chemistry that constitutes an open area of investigation (Fig. 1). First, persulfides are more acidic than thiols. This determines that the ionized species (RSS–) are highly available at physiological pH. Second, like thiols, persulfides are nucleophilic and can react with electrophiles (E+), although the products that are formed are different, disulfides in the case of persulfides and thioethers in the case of thiols. The ionized species are better nucleophiles than thiolates and have higher availability at physiological pH. Third, unlike thiols, which can only act as nucleophiles, persulfides, when protonated, also have electrophilic character, which can be located in both sulfur atoms. Fourth, they can react with oxidants. They are particularly good one-electron reductants, due in part to the relative stability of the RSS• radicals formed. Besides, persulfide formation protects protein thiols from irreversible oxidative damage, since the products of persulfide oxidation (RSSO2H and RSSO3H) can be reduced to thiols.
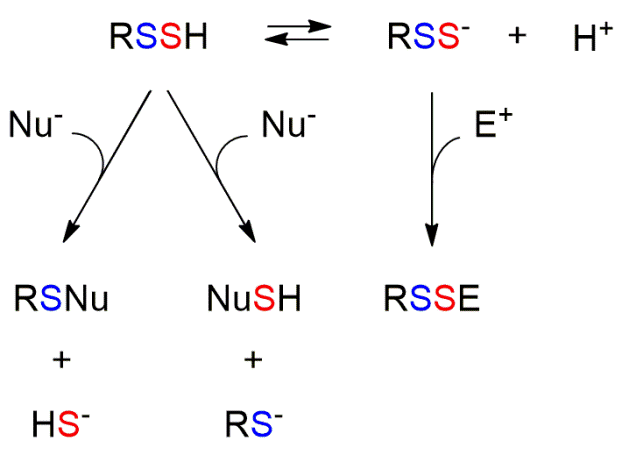
Fig. 1. Reactivity of persulfides
To sum up, persulfides are nowadays raising interest as candidate species for the transduction of the signals triggered by H2S. For further reading on H2S and persulfides, readers are referred to the recent reviews cited below and the references therein. It is likely that the next years will see an increase in the understanding of the biochemistry of these fascinating species.
Further reading
- M. R. Filipovic, J. Zivanovic, B. Alvarez, R. Banerjee. Chemical Biology of H2S Signaling through Persulfidation Chemical Reviews, 118(3): 1253–337, 2017. | doi: 10.1021/acs.chemrev.7b00205
- D. Benchoam, E. Cuevasanta, M. Möller, B. Alvarez. Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species Antioxidants, 8(2): 48, 2019. | doi: 10.3390/antiox8020048
- N. Lau, M. D. Pluth. Reactive sulfur species (RSS): persulfides, polysulfides, potential, and problems Current Opinion in Chemical Biology, 49: 1–8, 2019. | doi: 10.1016/j.cbpa.2018.08.012
- D. Benchoam, E. Cuevasanta, M. Möller, B. Alvarez. Persulfides, at the crossroads between hydrogen sulfide and thiols Essays in Biochemistry, 64(1): 155–68, 2020. | doi: 10.1042/ebc20190049
- J. M. Fukuto, V. S. Vega, C. Works, J. Lin. The chemical biology of hydrogen sulfide and related hydropersulfides: interactions with biologically relevant metals and metalloproteins Current Opinion in Chemical Biology, 55: 52–8, 2020. | doi: 10.1016/j.cbpa.2019.11.013


{kind=link}
Add new comment